The story of CDKL5 so far.....
The story of CDKL5 begins with Rett Syndrome, which was first described by Andreas Rett, an Austrian paediatrician. His original article “Uber ein zerebral-atrophisches Syndrome bei Hyperammonemie” was printed in 1966. However, it wasn't until the 1980's that the characteristics of the syndrome were established. Affected children typically have normal development for the first 6 to 18 months of life, followed by arrest and then regression of their development. A number of typical clinical features then develop including loss of motor and cognitive function, loss of communication ability, spinal problems (scoliosis), epilepsy and characteristic stereotypic hand movements. Subsequent studies showed that in the majority of cases, Rett Syndrome was caused by mutations in a gene on the X-chromosome, the so-called X-linked methyl-CpG-binding protein 2, otherwise known as the MeCP2 gene.
During the 1980’s and 1990’s reports appeared about children who had developed some, but not all of the features of Rett Syndrome. In particular, they didn't have the early period of normal development but they had developed epileptic seizures early in life. They were therefore diagnosed as having “Atypical Rett Syndrome".
In 1998, a novel gene was identified in the Xp22 region of the X-chromosome. A number of genetic conditions had previously been mapped to this region. The new gene was initially called the STK9 (Serine-Threonine Kinase) gene.
In 2003, a study was published that linked disruption in the STK9 gene to infantile spasms and mental retardation. The STK9 gene subsequently became known as CDKL5, which stands for Cyclin-Dependent Kinase-Like 5. As you might expect, there are 4 other CDKL’s (1-4), which all appear to have some sort of role in brain physiology and development, although exactly what they do is not entirely clear.
In 2004, independent case-studies were published by the then Dr. John Christodoulou and Dr. Vera Kalsceuer on mutations in CDKL5 producing severe neurodevelopmental disorder and mental retardation. These papers concluded that mutations in the CDKL5 gene gave rise to a phenotype (observable characteristics) that overlapped with Rett syndrome.

Further studies appeared from around the world including the UK, Italy, France, Spain, India, Brazil, China and Denmark. Although CDKL5 mainly affects girls, we know that boys can be affected as well. CDKL5 is now becoming recognised as a distinct condition in its own right, and a recent study has supported this view. There are a number of research units around the world studying the genetic, molecular biology and clinical aspects of CDKL5, for which we are all grateful. There is a deeply united group of parents, families and friends of affected children, who are using social media forums to give support and advice, especially to those with newly diagnosed children, and to raise awareness of this devastating condition. A nice review of CDKL5, from the first identification of CDKL5 was published from Italy in 2012 which you may find of interest.
Normal developmental milestones....
There are many sources from which you can learn about normal milestones during early development. I have summarised these below for the first 2 years of life.

What happens in CDKL5....
Children with CDKL5 often develop seizures in their few weeks or months of life. Most affected children are girls, and most will fail to develop their normal milestones. Some may have stereotypical hand movements and others may have cortical visual impairment. In order to understand more about the clinical features of CDKL5, an international database was established in 2012. The research group is based in Australia with funding from the IFCR. Some early results from the database were presented at The 3rd European Rett conference in Maastricht in October 2013.
In relation to seizures, the median time of onset was at 6 weeks of age and seizures occurred daily in 88% of cases. Anti-epileptic drugs were the commonest treatment with the use of 3 or more being the most frequent. Almost half of children had been on the ketogenic diet at some point although this had reduced to just under a third. Vagal nerve stimulation had been used in 23%. Epilepsy remains severe for most individuals with CDKL5.
Preliminary studies on development and gross motor function in 76 females and 21 males showed that 88% had impaired gross motor function, and milestones, when achieved, were delayed. Independent walking was seen in 17% of females and one male, while sitting was reported in 56% of females and 21% of males. Overall, males tended to be more severely affected than females.
Information was also presented in relation to gastro-intestinal (GI) problems. They reported on 83 individuals on who data was available. They found that the majority (86%) had had a history of GI problems at some point. The commonest was constipation (76%) followed by reflux (75%). A gastrostomy had been performed in 22 cases and 13 had had anti-reflux surgery. CDKL5UK also presented the results from a parent survey on the same subject. Of 90 responses about 75% reported reflux, 25% had a g-tube for feeding and about 20% had had a Nissen’s Fundoplication. Although only about 15% reported constipation (bowels open less than 3 times a week) at the time of the survey, nearly 50% were taking medication to support bowel function. Overall, the results between the 2 posters were fairly similar, which is not surprising, given that the populations being sampled may be similar.
In 2015, the same group published a more detailed study from the database on the attainment of various milestones (link to text) . Their paper consisted of a study of 109 females aged between 3 months and 29 years, and 18 males aged between 10 months and 22 years. Some results for the females are shown opposite.
For males, the numbers were less with only 6 achieving independent sitting, 2 standing and 1 walking. A quarter attained babble by 7 months of age and there was one boy who spoke single words at 3 years of age. Males were generally more severely impaired than females.
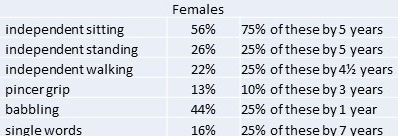
Although the authors noted some trends between genotype and phenotype, no clear relationship was established. The numbers in this study are still relatively small and larger studies will be required in the future to determine whether there is a genotype-phenotype relationship in CDKL5. This study does however suggest that there is variability within CDKL5 in terms of the attainment of developmental milestones and that there may be children whose presentation is different to the clinical picture described in earlier reports.
CDKL5UK has produced a Guide for Professionals that you might find of interest.