The genetics of CDKL5...
How genes work......
To understand how mutations cause conditions like CDKL5 it is worthwhile understanding the basics of how genes work. Genes are contained within our DNA. A gene is the "blueprint" for making a protein and is composed of a chain of bases or base-pairs. There are 4 bases used in DNA, Adenine, Cytosine, Guanine and Thymine. They are often referred to as base-pairs because in DNA they exist in pairs, Adenine always pairs with Thymine, and Guanine with Cytosine. This was one of the key findings that led to the discovery of the structure of DNA by Crick and Watson in 1953.
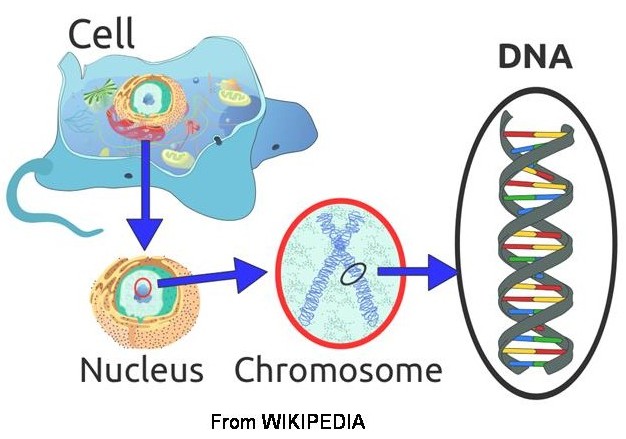
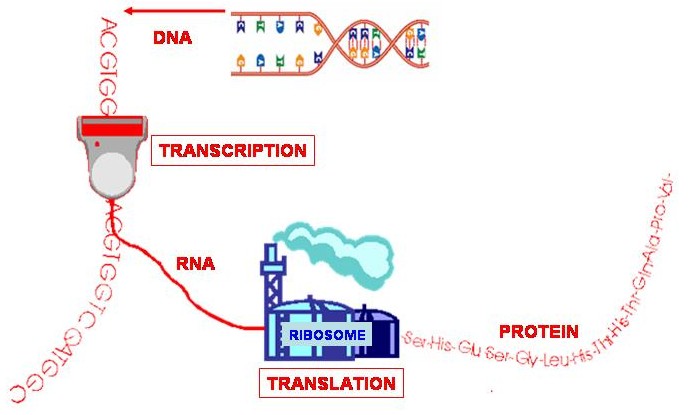
A gene is a specific section of DNA that codes for a protein. A protein consists of a chain of amino acids, and every 3 base-pairs, called a codon, in the gene code for an amino acid. The code is converted into a protein via RNA. If the gene is the "blueprint" then RNA is the "template" that is taken from the gene and used to make the protein.
So, the RNA template is taken from DNA, through a process called transcription. In RNA, Thymine (T) is replaced by Uracil (U). A structure called a ribosome (a sort of protein factory) then reads the RNA template and uses it to construct a chain of amino acids through a process called translation - and hey presto, you have a protein!
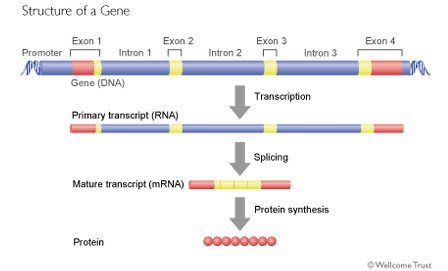
Just as a sentence is made up of words and spaces between the words, so a gene is made up of exons (the words) and introns (the spaces between). When the genetic code is being read and converted into a protein, the introns are removed and the exons then spliced together to produce the genetic code that will ultimately be read to make the protein.
What is interesting, however, is that the spaces between the words (introns) are actually much longer that the words (exons) themselves.
So a gene might look like this.
Furthermore, it may be that the introns actually contain important information about how the gene, or indeed another gene further along the DNA chain, is read and converted into a protein.
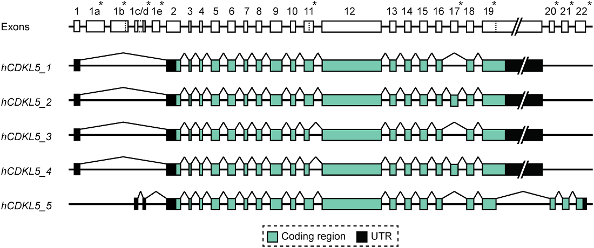
The CDKL5 Gene
The CDKL5 gene contains 24 exons. The 24 exons include exons 1, 1a and 1b which do not appear to contribute directly to the structure of the protein in that they are untranslated.
Exons 2 to 11 code for the kinase domain of the protein, and although there seems to be some evidence from the literature that mutations in this part of the gene might produce more severe phenotypes, the situation is far from clear. There are other mechanisms - so called epigenetic factors - that can potentially have a role to play in the severity of the phenotype produced.
Recent research by Hector et al has established that there are at least 5 distinct transcripts taken from the CDKL5 gene. They have also suggested a new way of labelling these various transcripts.
And so to mutations...
The order of the base-pairs is essential to the formation of the protein. A mutation can corrupt the order of the base-pairs so that the sequence of amino acids is wrong. Consequently, the protein won't be made properly and therefore won't function properly. The initial studies on girls with CDKL5 identified mutations defined as single base deletions (a base is effectively one letter in the sentence that is the gene code), and chromosomal translocations (a rearrangement of letters).
So, for example.....
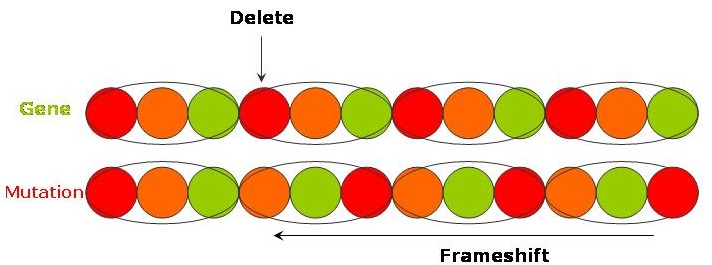
"The long and winding road" - a deletion of a causes everything after to shift along (a so called frameshift) and gives
"The long ndw indingr oad!"
"It's been a hard day's night and I've been working like a dog" - A translocation gives
"It's been a hard day's dog and I've been working like a night"
In both cases, the mutation has altered the order of the gene or sentence so that its intended meaning or function is lost.
Common mutations in CDKL5 are substitutions. So...
"Drive my car" might become "Drive my cat"
This is also known as a missense mutation, again because the structure and therefore function is altered.
Frameshifts and "Stop" codons...
As you saw in "The long and winding road" example above, if a mutation causes base-pairs to be deleted then all the subsequent bases will shift along - this is called a frameshift. The same occurs if there is an insertion. The offsetting of base-pairs will mean that new codons are being read with the result that the sequence of amino acids will be completely changed from what was originally coded.
A "Stop" codon is a codon that signifies that the end of the protein has been reached - and for obvious reasons is usually found at the end of the base-pair sequence coding for the protein. There are 3 stop codons - TAA, TAG and TGA. Because of the resulting frameshift, a deletion or insertion may produce a new codon that is a "Stop" codon. This might occur anywhere along the gene subsequent to the mutation and will cause the production of the protein to end prematurely resulting in a truncated protein.
If a substitution, as discussed above, directly produces a stop codon, then this is also known as a nonsense mutation. Other types of mutations are described here.
How are mutations described in reports....
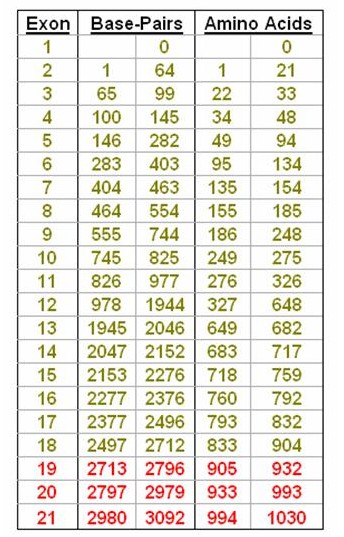
The CDKL5 gene has 21 exons containing 3092 base-pairs that code for 1030 amino acids, although it is now thought that the main CDKL5 protein is only coded by exons up to 18. Broadly speaking mutations are described using 2 formats although other forms are also used. As we have seen above, a mutation in a base-pair will cause a change in the corresponding amino acid and it is this change that affects the structure of the CDKL5 protein and therefore it's function. The report you receive may therefore refer either to which base is affected - in which case the mutation description begins with a "c." - or to which amino acid has consequently been changed - denoted by a "p.".
So, for instance, c.175C>T signifies that the base Cytosine at position 175 (which is in exon 5) has been replaced by Thymine. This is a substitution. Another example would be c.2047delG which is a deletion of the base Guanine at position 2047 (in exon 14). Another type of mutation is an insertion. So, c.865insA signifies that the base Adenine has been inserted into the CDKL5 gene at position 865 which is in exon 11. Note, the affected exon is not included in the format.
If the report is referring to the consequential change in amino acid, then you will see something like p.Ala40Val, which signifies that the amino acid Alanine has been replaced with the amino acid Valine at position 40 in the protein chain. Letters are also used to denote amino acids, so the same mutation might also be written as p.A40V. This change in amino acid from Alanine to Valine occurs because of a base substitution at position 119 in exon 4 - written as c.119C>T. Within a report, you may find that either one of both formats are used.
A truncated protein - due to either a nonsense mutation or deletion/insertion producing an early stop codon - is usually signified with an "X" as in p.R59X which is due to the substitution mentioned above, c.175C>T, which is also therefore a nonsense mutation.
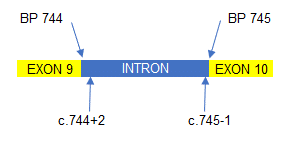
Mutations can also occur within the introns between each exon. So, in the example to the right, a mutation situated 2 base pairs (BP) after the end of exon 9 would be written as c.744+2 (which denotes the position of the mutation) followed by what the mutation was. A mutation towards the end of the intron, say 1 BP short of exon 10, would be written c.745-1 followed by the mutation.
How do mutations arise?
Broadly speaking there are two ways mutations can occur. Either they are acquired or inherited.
Acquired mutations
These mutations occur when genetic material is damaged at some point, usually during the cell cycle, where DNA is being copied prior to cell division. De novo mutations are those that occur for the first time and are not usually present in the parents of the affected child. The majority of CDKL5 mutations are assumed to arise this way. While historically it has been thought that de novo genetic disorders increased with the age of the mother, there is now some evidence that it is the father's age that might be more relevant to the development of some genetic diseases.
Inherited mutations
The inheritance patterns in autosomal conditions (involving chromosomes 1 to 22) are typically recognised as being dominant or recessive. The situation, however, is different in X-linked conditions such as CDKL5. There may ultimately be a number of ways that a CDKL5 disorder can be inherited, but a current view is that some CDKL5 mutations might be inherited as a result of germ line mosaicism.
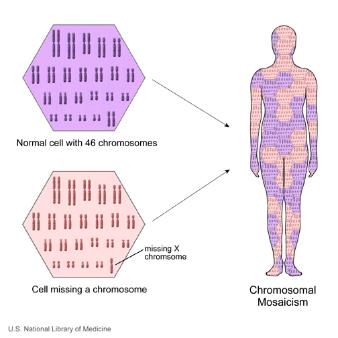
What is mosaicism?
The cells that make up the human body can broadly be divided into two sorts. The germ line cells, which are the sperm in males and eggs in females, while somatic cells include all the other remaining cells (which form muscles, bone, skin, brain etc..). Mosaicism occurs when a person has 2 cell populations each with distinct genetic information. So, one population contains “normal” genetic material, whilst the other population may have a mutation or other genetic abnormality. Mosaicism can affect both germ line cells and somatic cells.
Germ line mosaicism
In germ line mosaicism, the genetic abnormality is confined to a proportion of the germ line cells only, with the remainder being normal. In this situation, the individual will have no evidence of the underlying condition as the somatic cells that make up the rest of their body have normal genetic material. However, an individual can still pass the mutation on, through one of their abnormal germ cells. It has been suggested that this is a cause of CDKL5 disorders, perhaps in a very small number of cases.
Somatic mosaicism
There are a number of genetic disorders that display somatic mosaicism. The effects of somatic mosaicism are not usually passed on to offspring as the germ line cells are all normal. However, in some instances, individuals can have both somatic and germ line mosaicism, in which case the genetic abnormality can be passed on to offspring.
Finally, and very unusually, it would be possible for the mother to have a CDKL5 mutation but with an extremely skewed X-inactivation pattern (see below) such that the mutation was barely expressed. The mother might be relatively unaffected but she would effectively be a "carrier" of the CDKL5 mutation . This mechanism of inheritance would appear to be rare (I am not aware that it has yet been reported, at least not for CDKL5).
X-inactivation
The CDKL5 gene is located on the X chromosome. Although females have two X chromosomes (one from each parent), only one of them is needed for normal function, in fact, it would be detrimental for both chromosomes to be active together. Therefore, one of the X chromosomes is normally "switched off " through a process called X-inactivation.
The CDKL5 mutation is usually only present on one of the X chromosomes, so therefore, if it is the X chromosome with the mutation that is switched off, then it is possible to have the mutation and not have any effect at all as the X chromosome that remains active has a normal CDKL5 gene. However, it is not necessarily the same X chromosome that is inactivated in every cell in the body.
It turns out that the body can have groups of cells in which one particular X chromosome is inactive and other cells in which the other X chromosome is inactive. In the brain therefore, there can be areas where cells use the X chromosome with the mutation, and other areas where the cells are using the X chromosome with the normal CDKL5 gene.
Which particular X chromosome is inactivated appears to be random and down to "chance", but obviously, the greater proportion of cells using the chromosome with the mutation, the more severe the phenotype may be. Having said all that, it still remains to be established that the degree of X-inactivation does indeed affect the severity of the phenotype.
And so, a relationship between mutation and the severity of CDKL5 related symptoms remains unclear.
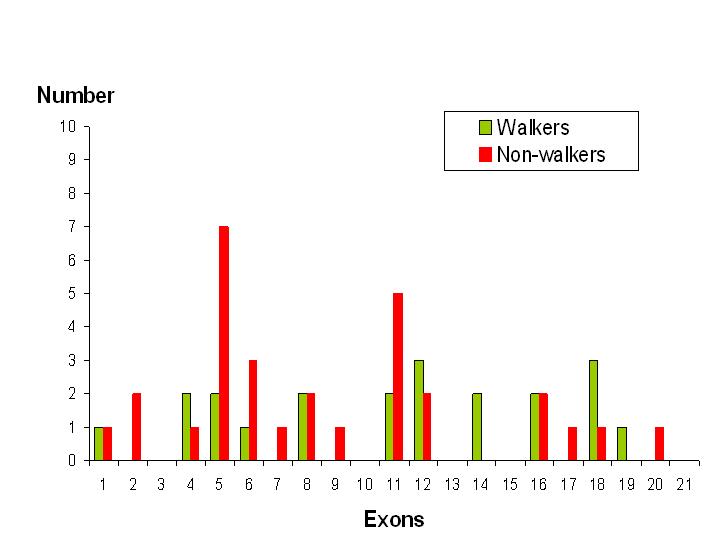
Interestingly, a review from France published in 2011, included a summary of the clinical details of 77 previously published cases of CDKL5. Of these, the best motor skills of 51 individuals are listed, of whom 21 appear to have various walking abilities.
Looking at walking ability in relation to the site of mutation, according to which exon is affected, a trend emerges.
Analysis of the data shows that only 30% of individuals with a mutation affecting exons 1 to 11 have some sort of walking ability whereas, that figure increases to 61% in those who have a mutation affecting exons 12 to 21.
This is obviously a relatively crude analysis and doesn’t take into account other factors such as the type of mutation, the degree of X-inactivation and multi-exon or intron involvement.
Other clinical factors may also be relevant such as the amount of therapy each individual has had or whether there are other orthopaedic issues such as hip or spine problems. Also, some relatively younger children may go on to develop an ability to walk whilst others who were walkers may lose their ability, perhaps because of poor epilepsy control. Furthermore, the numbers involved in this study are relatively small. Therefore, to help get answers to these questions a new CDKL5 Disorder International Registry Database has recently been developed. As more information about children with CDKL5 is recorded then the answers to many of these questions will hopefully become available.